Journal
of Medical Genetics 1988, 25, 827-830
|
John
Christodoulou, Roger K. Hall, Samuel Menahem, Ian J Hopkins, UND John G
Rogers Von den Abteilungen der Genetik, Zahnmedizin, Medizin und Neurologie, Königliche Kinderklinik, Parkville, Victoria 3052, Australien. |
| Zusammenfassung: Beschrieben
wird eine Familie mit sechs von einem Syndrom mit Epilepsie, Demenz und
Amelogenese imperfecta (Kohlschütter Syndrom) betroffenen Mitgliedern. Für diese
Störung wird ein autosomal rezessiver Erbgang angenommen. |
| Das
Kohlschütter Syndrom (McKusick No 22675) ist eine angeborene Störung, die durch
eine fortschreitende Demenz, zunehmende Spastik und Epilepsie charakterisiert
ist. Es ist verbunden mit einem angeborenen symmetrisch verlaufenden
Zahnschmelzdefekt, der sowohl das Milchgebiss als auch die bleibenden Zähne
betrifft, und der bewirkt, dass die Zähne dünne, gelbe und rauhe Kronen haben.
Es blieb unklar ob diese Störung x-chromosomal rezessiv oder autosomal rezessiv
gebunden ist. Wir beschreiben hier eine Familie, die deutlich einen autosomal
rezessiven Erbgang zeigt. |
|
Fallbeschreibung Abbildung
1 zeigt den Stammbaum dieser Familie.Es gibt sechs betroffene Personen. Das
erste Kind blieb normal und das siebte Kind ist sehr wahrscheinlich betroffen.
Die Eltern dieser Patienten sind ihrer Herkunft nach Sizilianer, und obwohl sie
aus der selben kleinen eng verflochtenen Stadt kommen (ungefähr 5000 Einwohner),
bestreiten sie eine Verwandtschaft. Vier mütterliche Verwandte (alle weiblich)
werden als geistig zurückgeblieben bezeichnet, aber der Zustand ihrer Zähne ist
nicht bekannt. Der väterliche Ast zeigt keine dentalen, neurologischen oder
andere relevante Anzeichen. IV.1
ist ein Mädchen mit normaler Entwicklung und normalem Intellekt und zeigt keine
Abnormalitäten im Zahnbereich. Sie ist jetzt 15 Jahre alt. |
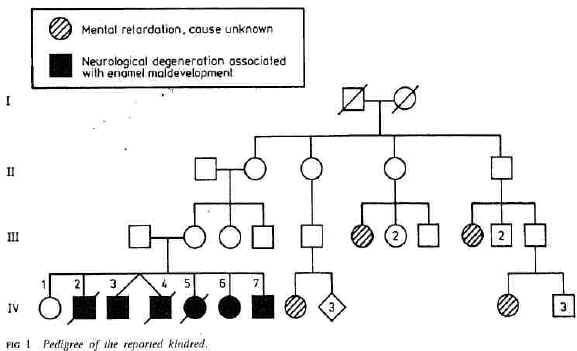
Abb. 1 |
|
IV.2
wurde zeitgemäss nach normaler Schwangerschaft geboren; unkomplizierte
Entbindung. Er entwickelte sich normal, bis er im Alter von 11 Monaten im Status
epilepticus ins Krankenhaus gebracht wurde. Trotz Behandlung mit verschiedenen
Antikonvulsiva, einschliesslich Phenytoin, Primidon und Nitrazepam schienne die
Anfälle extrem schwierig zu kontrollieren. Seine Entwicklung verlangsamte sich,
und wurde nach und nach rückläufig, und er ist jetzt schwer geistig behindert.
Im Alter von sieben Jahren besass er keine Sprachfähigkeit mehr und war nicht
imstande, selbständig zu essen, während er noch mit Unterstüzung laufen, und
ohne Hilfe sitzen konnte. Mit dem
Durchbruch der ersten Zähne im Alter von sechs Monaten, wurde eine
Zahnschmelzabnormalität bemerkt, jedoch ihre Bedeutung nicht erkannt. Er starb
im Alter von 10 ½ Jahren durch Lungenentzündung, Epilepsie und Abzehrung. Das
erste betroffene Kind wurde intensiven Untersuchungen unterzogen,
einschliesslich einer vollständigen Untersuchung des Blutes, Serum Sodium,
Potassium, Chloride, Calcium, Magnesium, Glukose, Röntgenaufnahme von Brust und
Schädel, die normal waren. Das EEG zeigte allgemein ausgeprägte slow wave
Aktivität, aber keine fokalen Auffälligkeiten. Die CSF Untersuchung zeigte bei
der ersten Vorstellung keine Abweichungen, lag hingegen in der Phase, die zum
Tod führte, bei einem erhöhten Laktatwert von 6.75 mmol/l (normaler Wert 1.04
bis 2.26). In der Endphase zeigte die CSF Aminosäureanalyse einen Glyzerinwert
von 18.2 umol/l (Normalwert 3.0 bis 10.2) und einen Alaninwert von 57.2 umol/l
(Normalwert 4.4. bis 42.0), während alle anderen Aminosäurekonzentrationen
normal waren. Suche nach Eiweiss und organischen Säuren im Urin blieben ohne
Abweichungen. Säure Base und Leberfunktionstest waren normal. Plasma C26:22 und
C24:22 langkettige Fettsäurewerte waren normal, ebenso wie die Lysosomal
Tätigkeit der weissen Blutkörperchen (einschliesslich ß-Galaktosidase,
Hexosaminidase A und B, Arylsulphatase A, Säure Phosphatase, ß-Glukuronidase,
alpha-Fukosidase, ß-Glukocerebrosidase, Sphingomyelinase, und
ß-Galaktocerebrosidase). Eine Biopsie des Bindegewebes zeigte keine
Einschlüsse. |
 |

Abb.2 Röntgenaufnahme des Gebisses von IV.6 im Alter von vier Jahren. Man beachte den dünnen, hypoplastischen Zahnschmelz der Molare und Spitzen (cuspids) des Milchgebisses und der ersten bleibenden Molare.
|
|
Es gab
fünf weitere betroffene Geschwister in dieser Familie, und ihre Klinik ist in
der Tabelle aufgeführt. Sie hatten alle einen normalen Grundstoffwechsel und
Urinwerte. IV.3 wurde zusätzlich eingehender interviewt. Diese Test beinhalteten
Fibroplastkulturen einer Reihe von Enzymen (Pyrovat Dehydrogenase, Cytochrom c
Oxydase, Succinatecytochrome c Reductase, NADH-Cytochrome c Reductase, und E1
ATPase), G gebundene Karyotype, Urinsäureserum, Porphyrin Blut Screen,
Blutammoniak, und CSF Laktat, Pyrovat und Aminosäuren. Alles ergab normale
Ergebnisse. Keins der Kinder hatte ein CT oder MRT des Gehirns. Das
Phenylalanin-Plasma der Mutter war normal. |

Abb.3 Das Erscheinungsbild der Zähne von IV.6 im Alter von fünf Jahren.
|
|
Diskussion Die
klinischen Syptome der betroffenen Geschwister dieser Familie gleichen sehr
stark der von Kohlschütter et al (1) beschriebenen Familie. In der von uns
beschriebenen Familie entwickelten sich die betroffenen Kinder bis zum Beginn
der Anfallstätigkeit, im Alter von sieben bis zweiundzwanzig Monaten, normal.
Bei den drei ältesten Kindern.verlangsamte sich die psychiomotorische
Entwicklung und wurde nach und nahc rückläufig, was zu schwerer geistiger
Behinderung in der Kindheit führte. Es gab Schwierigkeiten die Anfälle zu
kontrollieren. Alle
betroffenen Kinder zeigten den als Amelogenese imperfecta bekannten
Zahnschmelzdefekt, während das einzige normale Kind und die Eltern keine
Abweichungen der Zähne zeigen. Drei der sechs betroffenen Kinder sind gestorben.
Bis auf die Zahnfehlbildungen wurden keine Missbildungen bei den erkrankten
Kindern gefunden. Der
geistig retardierten Vorfahren, die im Stammbaum gezeigt werden, leben in
Italien, und über sie sind keine Informationen verfügbar. Biochemische
und zytogenetische Untersuchungen bei einigen betroffenen Personen gaben keine
Hinweise auf eine zugrundeliegende Stoffwechselstörung. Die erhöhten CSF
Laktatwerte des ältesten Jungen konnten keiner Stoffwechselstörung zugeordnet
werden. Analysen
früherer Stammbäume ergaben keine eindeutigen Hinweise. Dies spricht stark für
einen autosomal rezessiven Erbgang. Sowohl männliche als auch weibliche
Nachkommen sind gleichermassen schwer betroffen, während die Eltern klinisch
unauffällig sind. Trotz des ihnen vor Augen geführten Risikos haben die Eltern
beschlossen weitere Kinder zu bekommen, da sie auf einen gesunden Sohn
hoffen. Das
deutlichste physische Charakteristikum dieser Erkrankung ist der
Zahnschmelzdefekt, der es ermöglichte, das Syndrom zu diagnostizieren. Eine
normaleZahnschmelzentwicklung beeinhaltet die Formation und Absonderung einer
organischen Matrix, die die Proteine Amelogenin und Enamelin in sich trägt, und
die für die Entwicklung des ersten Gebisses in der siebten Schwangerschaftswoche
beginnt. Hierauf folgt die Mineralisierung der Matrix in der neunten Woche und
anschliessend die Ausreifung des Zahnschmelzes unter Wandlung von Amelogenin zu
Enamelin. Der hier beschriebeneZahnschmelzdefekt, bekannt als Amelogenese
imperrfecta ist das Ergebnis eine Störung auf der Ebene der Organmatrix und
zieht sich durch bis zur Calcifizierung. Diese Störung tritt mit einer
Häufigkeit von 1 : 4000 bis 1: 14000 auf. Amelogenese imperfecta tritt als
einzelnes Symptom sowohl in autosomal als auch x-chromosomal gebundenen
Erbgängen auf, und zwar sowohl dominant als auch rezessiv in jeder Gruppe.
Dieser Typus eines Zahnschmelzdefektes, der alle Zähne sowohl des Milch- als
auch des bleibenden Gebisses betrifft, aber von der Entwicklung her nicht
zeitgebunden ist, wie bei hypoplastischen Zahnschmelzdefekten gesehen, kann als
isolierter Defekt auftreten, wurde aber niemals bei einem anderen als dem
Kohlschütter Syndrom beschrieben. Kohlschütter
et al (1) nahmen an, dass es sich um ein einzelnes defektes Gen handle, das für
die Erregbarkeit der Haut (membran excitability), Zahnschmelzbildung und
vielleicht Schweissproduktion zuständig ist. Amelogenese imperfecta könnte das
Ergebnis einer Veränderung im Enamelin sein. Das ursprüngliche Gewebe, aus dem
der Zahnschmelz entsteht, ist vom Ursprung her ektodermal, genauso, wie das
Gehirn.(6). Viele Zustände, die neurologische Defekte zeigen, haben
Zahnabnormalitäten als Begleiterscheinungen (7). Die Hypothese liegt nahe, dass
die hier beschriebene Störung auf einen Defekt bei der Ausreifung des
ektodermalen Gewebes zurückgeht. |
|
Literatur 1
Kohlschütter A., Chappuis D, Meter C, Tönz O, Vassella F, Herschkowitz N.
Familial epilepsy and yellow teeth – a disease of the central nervous system
associated with enamel hypoplasia. Helv Paediatr Acta 1974; 29:283-94 2
Witkop CJ Jr, Sauk JJ Jr, Heritable defects of enamel. In: Stewart RE, Prescott
GH, eds. Oral facial genetics. St Louis: Mosby, 1976:151-226. 3
Wright JT, Analysis of a kindred with amelogenesis imperfecta. J Oral Pathol
1983; 14:366-74 4
Sundel S, Koch G,. Hereditary amelogenesis imperfecta. !.Epidemiology and
clinical classification in a Swedish child population. Swed. Dent J 1983; 9:
157-69. 5
Menanteau J, Mitre D, Raher S. An in vitro study of enamel protein degradation
in developing bovine enamel. Arch Oral Biol. 1986; 12:807-10. 6
Moore KL, The skin, cutaneous appendages and teeth. In: The developing human.
Philadelphia: Saunders, 1982:228-31. 7
Smith DW. Appendix. Pattern of malformation differential diagnosis by anomalies.
In: Recognizable patterns of human malformations. Philadelphia: Saunders,
1982:27-8. |
|
Schriftverkehr und Nachdruckgenehmigungen bei Dr.John Christodoulou, Department of Genetics, Royal Children’s Hospital, Flemington Road, Parkville, Victoria 3052, Australien. |
www.heininfo.de -
(c) vom
24.10.2012